从零开始设计分子是化学中最难的问题之一。这不仅仅是关于知道连接哪些原子——而是关于知道反应的正确顺序,何时保护分子的敏感部分,以及如何避免可能破坏数月实验工作的死胡同。
传统上,这些知识存在于经验丰富的化学家头脑中。现在,洛桑联邦理工学院的一个团队想将其放入一个语言模型中。
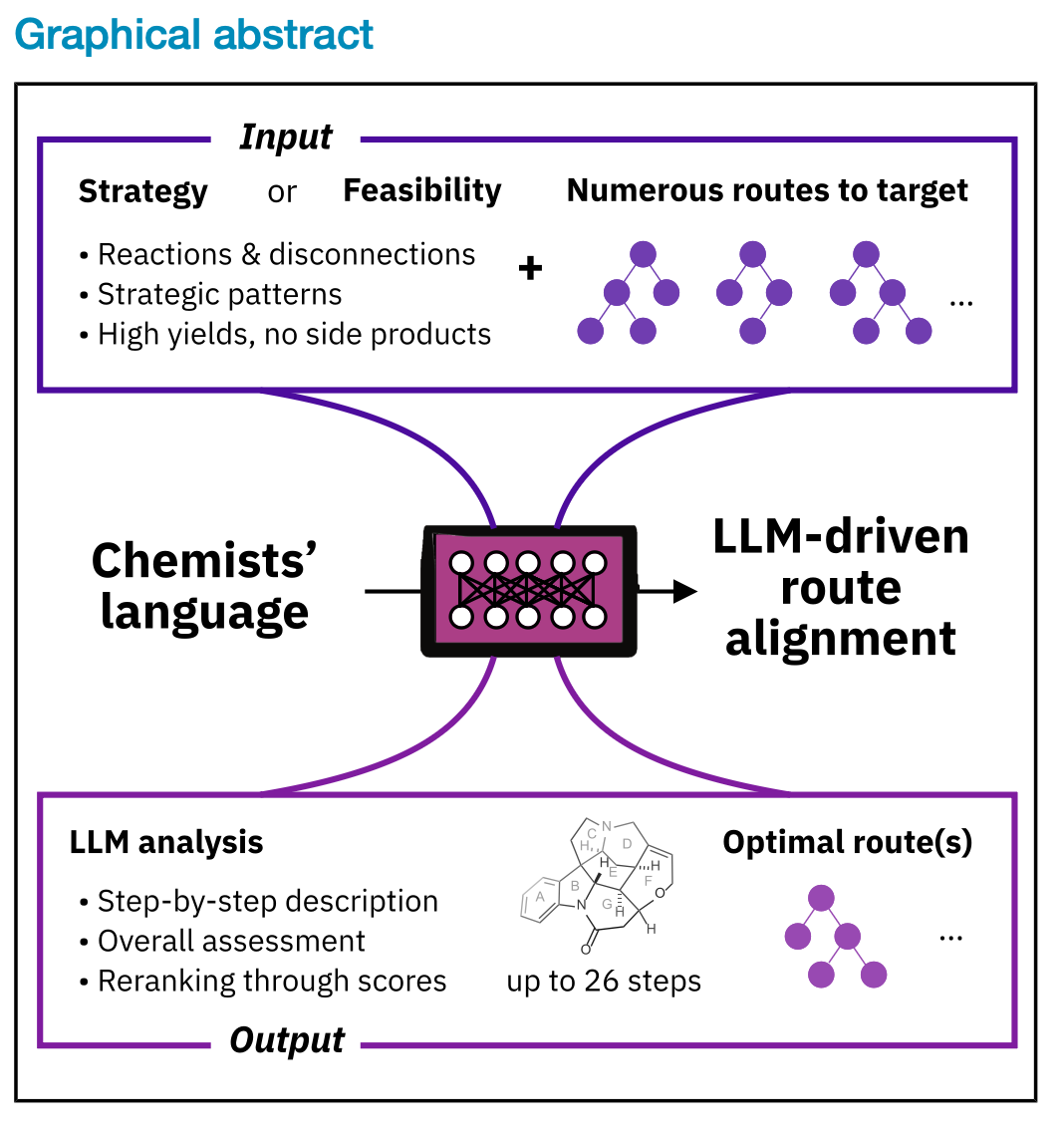
由菲利普·施瓦勒领导的研究人员本周在《Matter》上发布了一篇论文,描述了Synthegy,这是一个将大型语言模型作为化学合成规划推理引擎的框架。关键的洞察虽然微妙但重要:团队不是要求人工智能生成分子,而是利用人工智能评估传统软件已经产生的合成路线。
它的工作原理如下:化学家用简单的英语输入一个目标,比如“在早期阶段形成嘧啶环”。现有的逆合成软件——它通过将目标分子分解为更简单的部分来工作——然后生成几十个或几百个可能的合成路线。
Synthegy将每条路线转换为文本并交给大型语言模型(LLM),该模型对每条路线与化学家的指示的匹配程度进行评分。最好的路线会被筛选出来,并附有解释理由的文字说明。
研究的首席作者安德烈斯·M·布兰在洛桑联邦理工学院的一份声明中表示:“在为化学家制作工具时,用户界面非常重要,而以前的工具依赖于繁琐的过滤器和规则。”
该系统在一项双盲研究中得到了验证,涉及36名独立的化学家审查368对路线。他们的选择与Synthegy的匹配率达到71.2%,这个数字大致与专家化学家之间的一致性相当。高级研究人员(教授和研究科学家)的认可率高于博士生,这表明该系统捕捉到了随着经验而来的相同战略直觉。
研究人员测试了多个人工智能模型,包括GPT-4o、Claude和DeepSeek-r1。多年来,人工智能在药物发现中取得了进展,但大多数方法集中于为特定任务训练的狭义模型。Synthegy旨在模块化——它可以与任何逆合成引擎的后端连接,以及与任何具备推理能力的LLM连接。在基准测试中,Gemini-2.5-pro得分最高,而DeepSeek-r1似乎是一个强大的开源替代方案,可以在本地运行。
该框架还解决了第二个问题:反应机制的阐明。这是关于为什么发生化学反应的问题——每一步中发生了什么电子运动。Synthegy将反应分解为基本动作,并让LLM评估每个候选步骤的化学合理性。在核亲电取代这样简单的反应中,最佳模型达到了近乎完美的准确率。
潜在的应用案例非常广泛。药物发现显然是一个明显的领域。人工智能已经显示出希望,能够预测癌症治疗结果,但同样的思路适用于任何化学家需要设计新材料或优化工业反应的地方。一个实际的细节是:使用Synthegy评估60条候选路线大约需要12分钟,费用大约为2到3美元的API费用。
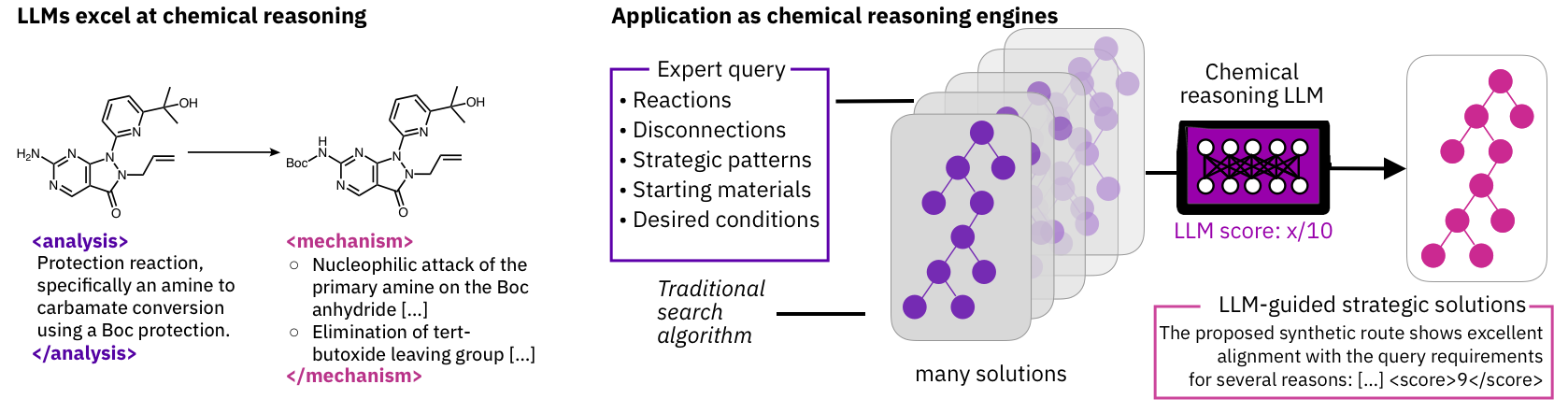
论文承认了当前的局限性。LLM有时会在其文本表示中误解反应的方向,导致错误的可行性判断。较小的模型表现得并没有比随机猜测更好。超过20步的路线难以连贯跟踪。
代码和基准测试在github.com/schwallergroup/steer上公开可用。
免责声明:本文章仅代表作者个人观点,不代表本平台的立场和观点。本文章仅供信息分享,不构成对任何人的任何投资建议。用户与作者之间的任何争议,与本平台无关。如网页中刊载的文章或图片涉及侵权,请提供相关的权利证明和身份证明发送邮件到support@aicoin.com,本平台相关工作人员将会进行核查。






